Экстракционная система имеет только одну степень свободы при Р = const и Т = const, т.е. произвольно можно менять только концентрацию вещества в одной фазе, концентрация же этого вещества во второй фазе оказывается величиной зависимой. Пусть одной фазой будет насыщенный раствор йода в воде, а второй фазой – органический растворитель (например, бензол). Если обе фазы привести в соприкосновение (перемешать) и датъ установиться равновесию, то часть йода из водного раствора перейдет в бензол.
Условия фазового равновесия при распределении одного компонента между двумя фазами записывается в виде:
К = ![]() .
.
Величина K называется константой распределения, если компонент А как в органической, так и в водной фазах находится в одинаковой химической форме.
Процесс экстракции можно записать в виде химического уравнения:
![]() .
.
Константа равновесия этого гетерогенного равновесия называется константой экстракции и записывается так:
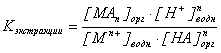 .
.
Нас будет интересовать не столько техника выполнения процесса, сколько движущая сила процесса. Какие причины заставляют вещество переходить из водной фазы в органическую?
Рассмотрим следующую гетерогенную систему:
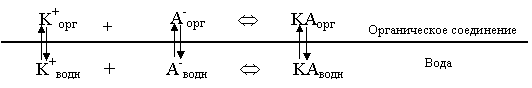 Здесь мы предполагаем, что все частицы, имеющиеся в водной фазе, могут, в принципе, переходить в органическую. Запишем выражения для констант распределения:
Здесь мы предполагаем, что все частицы, имеющиеся в водной фазе, могут, в принципе, переходить в органическую. Запишем выражения для констант распределения:
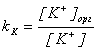 ,
, 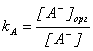 ,
, ![]() .
.
Если ![]() , мы имеем катионный вид экстракции. В органическую фазу переходит катион, на границе раздела фаз появляется заряд (в органическом растворителе положительный, в воде отрицательный). Для компенсации заряда органической фазы в нее переходит анион, и в целом система оказывается электронейтральной. Соединения КА в водной фазе может и не быть вовсе, если же оно имеется, то просто является поставщиком ионов.
, мы имеем катионный вид экстракции. В органическую фазу переходит катион, на границе раздела фаз появляется заряд (в органическом растворителе положительный, в воде отрицательный). Для компенсации заряда органической фазы в нее переходит анион, и в целом система оказывается электронейтральной. Соединения КА в водной фазе может и не быть вовсе, если же оно имеется, то просто является поставщиком ионов.
Если ![]() , мы имеем анионный вид экстракции. Механизм перехода вещества в органическую фазу такой, как и в случае катионной экстракции, только движущей силой процесса является переход аниона. Естественно, что при этом заряд на поверхности раздела фаз будет обращенным.
, мы имеем анионный вид экстракции. Механизм перехода вещества в органическую фазу такой, как и в случае катионной экстракции, только движущей силой процесса является переход аниона. Естественно, что при этом заряд на поверхности раздела фаз будет обращенным.
Если ![]() и
и ![]() , то мы имеем ассоциативный вид экстракции. Движущей силой процесса является переход соединения KA в органическую фазу. Ионы K+ и А‑ в водной фазе служат только материалом для создания соединения. В органической фазе K+ и А‑ могут появиться как продукты диссоциации соединения КА.
, то мы имеем ассоциативный вид экстракции. Движущей силой процесса является переход соединения KA в органическую фазу. Ионы K+ и А‑ в водной фазе служат только материалом для создания соединения. В органической фазе K+ и А‑ могут появиться как продукты диссоциации соединения КА.
Каким бы ни был вид экстракции, с энергетической точки зрения направление процесса определяет разность между энергией гидратации частицы молекулами воды и энергией сольватации этой же частицы молекулами органического растворителя.
С этой точки зрения интересно рассмотреть экстракцию минеральных кислот. Как правило, эти кислоты относятся к сильным или, в крайнем случае, к кислотам средней силы. В водных растворах они существуют в виде гидратированного катиона гидроксония и анионов, гидратированных в меньшей степени, поскольку при равном по величине заряде они имеют значительно больший радиус по сравнению с протоном. Небольшие ионные формы сильно гидратированы высокополярными молекулами воды, поэтому прочно удерживаются в водной фазе.
Энергия сольватации этих частиц неполярными молекулами органического растворителя в несколько раз меньше и не может скомпенсировать потерю энергии при дегидратации. Поэтому такими растворителями минеральные кислоты не экстрагируются, либо экстрагируются в чрезвычайно малой степени. Для того чтобы экстракция протекала в заметной степени, необходимы более полярные растворители, способные достаточно эффективно сольватировать ионные формы. Такими растворителями являются: простые эфиры, кетоны, сложные эфиры и спирты. Функциональные группы этих соединений сообщают исходным молекулам основные свойства, благодаря этому возрастает их способность сольватировать частицы кислотного характера, и эффективность их как экстрагентов возрастает.
Наибольшей основностью обладают амины, которые могут быть первичными, вторичными и третичными: RNH2, R2NH, R3N. Основные свойства молекуле придает неподеленная пара электронов азота. Взаимодействием этого центра и объясняется высокая сольватирующая способность этих частиц по отношению к частицам кислотного характера. Алифатические радикалы R делают вещество нерастворимым в воде и не способным гидратироваться молекулами воды; сольватация же неполярными молекулами растворителя наоборот возрастает. Поэтому неудивительно, почему амины экстрагируют минеральные кислоты с большими коэффициентами распределения.
В двухфазной системе, содержащей ионы, способные сольватироваться, органический растворитель может конкурировать с водой за координационные места у этих ионов. Если растворитель обладает подходящими свойствами, он выходит победителем в этом процессе, и образуются соединения, которые достаточно сольватированы неводным растворителем, что и заставляет их переходить в водную фазу.
Водородный ион обладает свойствами, которые делают его особенно удобным для такой координационной сольватации. Его уникальное поведение в водном растворе определяется высокой плотностью заряда. В настоящее время экспериментально
доказано, что в воде ион Н3О+ окружен в первом гидратном слое тремя молекулами воды, которые связаны с ним водородными связями, образуя симметричную структуру. Положительный заряд делокализован на трех центральных атомах водорода.
Экстракция минеральных кислот протекает по катионному механизму, однако это вовсе не означает, что анион кислоты никак не влияет на коэффициент распределения. Если мы возьмем кислоты: HClO4, HI, HBr, HCl, H2SO4, H3PO4 и будем их экстрагировать каким-либо основным растворителем, например, сложным эфиром в керосине, то оказывается, что коэффициенты распределения по своим значениям расположатся в следующем порядке:
HClO4 > HI > HBr > HCl > H2SO4 > H3PO4,
причем две последние кислоты практически не будут экстрагироваться.
Поскольку катион один (ион гидроксония), различия в значениях коэффициентов распределения можно объяснить единственным способом – различной энергией гидратации анионов. Это вполне понятно: при одинаковом заряде энергия гидратации убывает с увеличением радиуса аниона, а при одинаковом радиусе увеличение заряда приводит к росту энергии гидратации.
При рассмотрении экстракции минеральных кислот не следует упускать из виду обстоятельство, что сильная в водных растворах кислота при переходе в органическую фазу с малой диэлектрической проницаемостью может оказаться слабой. Экстракция будет протекать по ассоциативному типу, т.е. распределяться между фазами будут недиссоциированные молекулы кислоты. При этом могут образоваться соединения между молекулой экстрагента и недиссоциированной молекулой кислоты.
На основании всего изложенного можно сформулировать следующие соображения для объяснения порядка экстракции различных кислот. По-видимому, можно рассматривать, по крайней мере, три фактора:
1) силу кислоты. В настоящее время кислоты подразделяются на две группы: сильные кислоты и слабые кислоты. Следует ожидать, что кислоты второй группы лучше экстрагируются неполярными растворителями, так как они легче образуют нейтральные молекулы;
2) размер молекулы. Кислоты с большими анионами лучше экстрагируются неполярными и основными растворителями;
3) сольватацию. При использовании растворителей, не содержащих гидроксилов, анионы не могут сольватироваться в первой координационной сфере. Однако в водных растворах они могут быть гидратированы. Так, по экспериментальным данным галогениды в водной фазе связаны прочнее, чем перхлорат. Сочетание этого фактора с двумя первыми обеспечивает следующий порядок экстракции:
CCl3COOH > HNO3 > HClO4 > HI > HBr > HCl > H2SO4 > H3PO4.
В настоящее время экстракционные системы принято квалифицировать по типу соединения, извлекающегося в органическую фазу. Наиболее простым случаем является распределение простых ковалентных молекул. Упоминавшаяся нами экстракция йода является типичным представителем этого класса. Коэффициент распределения в этом случае равен константе распределения и равен отношению растворимостей распределяющегося вещества в органической и водной фазах.
Следует заметить, что для расчета коэффициента распределения нужно брать растворимости в фазах, взаимно насыщенных друг другом. Например, в системе:
вода – четыреххлористый углерод – йод
используется растворимость йода в воде, насыщенной четыреххлористым углеродом и растворимость йода в четыреххлористом углероде, насыщенном водой. Отношение растворимостей в этой системе равно 89,6, а константа распределения, определенная независимым методом – 89,9. Совпадение прекрасное!
Величина константы распределения незначительно зависит от концентрации йода, что является следствием использования концентраций вместо активностей. Температурная зависимость константы распределения такая, какую следует ожидать, учитывая теплоты растворения йода в четыреххлористом углероде и воде. Разность составляет (+770 ± 480) кал/моль. Экспериментально найденная теплота экстракции равна (+490 ± 25) кал/моль. Учитывая ошибки в определении этих величин, можно сказать, что совпадение вполне удовлетворительное.
Таким образом, для систем, в условиях, близких к идеальным, главной причиной экстракции ковалентного соединения в органическую фазу и главным фактором, определяющим численное значение константы распределения, является различие в растворимости. Взаимодействие экстрагируемого вещества с водой может быть сравнимо с взаимодействием с органическим растворителем или даже сильнее его. Но взаимодействие между молекулами воды благодаря сильным водородным связям всегда значительно сильнее взаимодействия между молекулами органического растворителя, и поэтому водная фаза как бы стремится вытолкнуть ковалентную молекулу в органическую фазу.
Вода, как сейчас достоверно известно, имеет структуру, характеризующуюся наличием ближнего порядка и отсутствием дальнего. В этой структуре имеются полости, в которых размещаются молекулы растворенного вещества. Естественно, что размер полостей должен быть больше размеров молекул растворенного вещества, чтобы сохранялась первоначальная структура воды, в противном случае либо структура должна разрушиться с затратой энергии, либо вещество будет вытолкнуто в другую жидкую фазу, где таких ограничений нет.
Из этих представлений следует, что размер молекулы экстрагируемого вещества должен влиять на его экстракцию. На влияние размера молекул указывают также данные о распределении различных органических веществ между водой и диэтиловым эфиром. Было установлено, что экстракция повышается в два – четыре раза на каждую новую группу СН2, введенную в молекулы соединений гомологического ряда.
Не следует думать, что в случае распределения ковалентных молекул коэффициент распределения всегда равен константе распределения. Он зависит от состава как водной, так и органической фаз, а также от величин констант равновесия процессов, протекающих в этих фазах. И только в определенном случае, когда химического взаимодействия в фазах нет, коэффициент распределения будет равен константе распределения.
Наибольшей сложностью в экстракционном поведении отличаются системы комплексных металлсодержащих кислот. Эти соединения имеют общую формулу:
НхМеХу,
где х = 1 или 2, а у = 4 или 6.
Большинство элементов периодической таблицы образует такие соединения, когда в качестве X выступают галогенид-ионы или псевдогалогенидные ионы, например, СN- и SCN-. Для этих кислот характерны все свойства простых сильных кислот, но в этом случае картина экстракции усложняется тем, что экстрагируемый комплекс должен быть образован в результате реакции катиона металла с соответствующим числом галогенидных ионов. Экстракция, как правило, проводится при высоких концентрациях галогеноводородной кислоты, которая одновременно экстрагируется в органическую фазу.
Рассмотрим этот случай на примере экстракции хлорного железа (рис. 7.3). Экспериментальные данные по изучению экстракции предусматривают получение зависимости коэффициента распределения от состава водной фазы.
С ростом концентрации кислоты коэффициент распределения растет, причем для диэтилового и диизопропилового эфиров он достигает максимума и затем начинает
уменьшаться. А для b,b’-дихлордиэтилового эфира максимума нет – коэффициент распределения растет непрерывно вплоть до 10 М соляной кислоты.
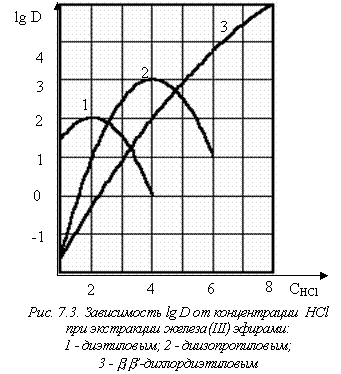 Первоначальное объяснение такого изменения коэффициента распределения заключалось в предположении, что в органическую фазу извлекается нейтральная частица FeСl3, концентрация которой растет в водной фазе по мере увеличения концентрации HСl. Падение коэффициента распределения при высоких кислотностях связывалось с образованием анионных частиц типа FeСl4-, которые не переходят в органическую фазу. В этом объяснении оставалось непонятным, почему у одних экстрагентов наблюдается максимум, а у других нет, к тому же, даже если максимумы имеются, они лежат при разных кислотностях водной фазы.
Первоначальное объяснение такого изменения коэффициента распределения заключалось в предположении, что в органическую фазу извлекается нейтральная частица FeСl3, концентрация которой растет в водной фазе по мере увеличения концентрации HСl. Падение коэффициента распределения при высоких кислотностях связывалось с образованием анионных частиц типа FeСl4-, которые не переходят в органическую фазу. В этом объяснении оставалось непонятным, почему у одних экстрагентов наблюдается максимум, а у других нет, к тому же, даже если максимумы имеются, они лежат при разных кислотностях водной фазы.
По результатам анализа экстрактов в условиях насыщения органической фазы было установлено, что экстрагирующееся соединение отвечает составу: НFеСl4.4,5H2O. Из концентрированных растворов соляной кислоты извлекается продукт с отношением Сl : Fe > 4, количество же экстрагированной воды всегда лежит в интервале 4 – 5 молекул на 1 г/атом железа.
Интересно отметить, что при экстракции железа b,b’-дихлордиэтиловым эфиром отношение Сl : Fe ни при каких концентрациях HСl не превышает 4. Наличие воды в экстракте стало понятным после установления состава экстрагирующегося соединения. Как всякая сильная минеральная кислота, это соединение переходит в органическую фазу по катионному механизму, причем катионом является гидратированный ион оксония, который и увлекает воду в органическую фазу.
Исследование спектров поглощения экстрактов растворов хлорного железа в концентрированной соляной кислоте и тонких слоев твердой соли KFeCl4 показало, что все спектры совершенно идентичны, а это значит, что ни гидратации, ни сольватации комплексного аниона FeCl4- в первой координационной сфере не происходит. Спектры поглощения аниона FeCl4- как в твердом состоянии, так и органической фазе не зависят от природы катиона, а это значит, что протон не связан химической связью с анионом в органической фазе.
Исследование электропроводности экстрактов показывает, что органические фазы слабо проводят ток, но, тем не менее, электропроводность экстрактов железа значительно больше, нежели электропроводность экстрактов НСl той же концентрации. Следовательно, кислота НFеСl4 сильнее соляной кислоты в органических фазах.
Теперь, зная, что представляет собой экстрагируемое соединение, можно по-другому взглянуть на форму кривой экстракции железа диэтиловым эфиром. Крутой подъем значений коэффициента распределения с увеличением исходной концентрации соляной кислоты в водной фазе в значительной мере обусловлен возрастанием доли железа, переходящего в экстрагируемый комплекс FеСl4- с увеличением концентрации хлорид-иона. Частично подъем определяется уменьшением активности воды с повышением концентрации HСl. Дело в том, что в концентрированных растворах почти все
имеющиеся молекулы воды связаны с ионами или молекулами, и свободных, несвязанных молекул воды нет; в этом случае говорят, что активность воды падает.
Уменьшение активности воды создает благоприятные условия для сольватации частиц молекулами органического растворителя, что и приводит к росту экстракции. Уменьшение активности воды лежит в основе эффекта высаливания. Этот эффект заключается в следующем: если к экстракционной системе добавить концентрированный раствор соли, не вступающей ни в какие реакции с компонентами системы, то коэффициент распределения экстрагирующегося вещества увеличивается. В случае экстракции железа такое высаливающее действие оказывает возрастающая концентрация соляной кислоты.
Максимум на кривой экстракции невозможно объяснить образованием анионных форм железа с высоким зарядом, поскольку формы FеСl52- и FеСl63- ни в водных, ни в органических растворах обнаружены не были. Дело здесь обстоит гораздо проще. С увеличением концентрации НСl в водной фазе эфир начинает во все большей степени переходить в воду, сольватируя здесь ион гидроксония.
В концентрированных растворах НСl в водную фазу переходит настолько много эфира, что происходит заметное изменение объемов фаз, и концентрация НСl в водной фазе уменьшается за счет разбавления. Действительно, равновесная концентрация НСl имеет максимум при 7 M исходной концентрации НСl в водной фазе, а затем уменьшается при более высоких исходных концентрациях кислоты.
Таким образом, если бы экстрагент абсолютно не растворялся в водной фазе при возрастании концентрации кислоты, максимума не было бы, что мы и наблюдаем у b,b’-дихлордиэтилового эфира. Из трех рассмотренных нами экстрагентов наибольшая растворимость – у диэтилового эфира, а наименьшая – у b,b’-дихлордиэтилового эфира.
Влияние большинства факторов на экстракцию железа можно выразить в количественной форме:
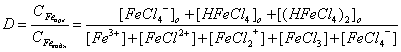 . (7.3)
. (7.3)
Константы распределения иона FeCl4- и H+ равны:
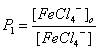 и
и ![]() .
.
Для процесса комплексообразования Fe3+ с хлорид-ионом можно записать ряд уравнений:
![]() ,
, ![]() ,
, ![]() ;
;
![]() ,
, ![]() ,
, ![]() ;
;
![]() ,
, ![]() ,
, ![]() ;
;
![]() ,
, ![]() ,
, ![]() .
.
Эти процессы идут в водной фазе, гидролизом и полимеризацией мы пренебрегаем, поскольку экстракция проводится из сильно кислых сред.
В органической фазе идут процессы образования ионных пар и их димеризация:
![]() ,
, ![]() ,
, ![]() ;
;
![]() ,
, ![]() ,
, ![]() .
.
Из константы распределения находим:
![]() ;
;
![]() ;
;
![]() .
.
Заменим в выражении (7.3) для коэффициента распределения концентрации различных равновесных форм через разновесные концентрации в водной фазе.
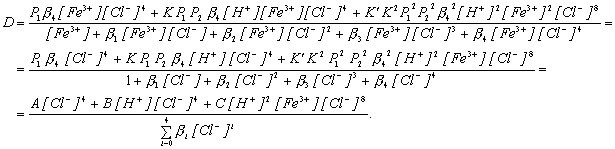
Из этого выражения видно, что коэффициент распределения железа сильно зависит от равновесной концентрации хлорид-иона в водной фазе, в несколько меньшей степени от равновесной концентрации ионов водорода в водной фазе и совсем слабо от концентрации ионов железа. В этой форме записи не нашло отражение изменение активности воды. Если бы мы пользовались не концентрациями, а активностями (a = c · g), то изменение активности воды вошло бы в множитель, содержащий все коэффициенты активности.
В растворителях с низкой диэлектрической проницаемостью ионные формы полностью отсутствуют, поэтому первым слагаемых числителя в полученном выражении можно пренебречь. Если использовать очень низкие концентрации, процессом димеризации в органической фазе также можно пренебречь, тогда
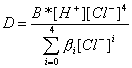 .
.
Как видно, коэффициент распределения в этом случае линейно зависит от концентрации ионов водорода и от концентрации ионов хлора в четвертой степени. Поскольку значение коэффициента распределения зависит от константы образования экстрагирующегося комплекса, то различные металлы будут иметь максимум экстракции при различных концентрациях соляной кислоты, и, тем самым, появляется возможность их разделения за счет изменения кислотности водной фазы, подобно тому, как это делается при осаждении сульфидов.
Например, при низких кислотностях (~1 M HCl) можно извлечь все золото, не затрагивая железа и сурьму, при средних кислотностях (4-8 M HCl) довольно эффективно отделить железо, а в концентрированных растворах (12 M HCl) экстрагировать сурьму.
Анион комплексных металлсодержащих кислот не гидратирован и не cольватирован в первой координационной сфере металла. В водных растворах он гидратируется за счет электростатических сил и, таким образом, энергия этого процесса зависит от размера и заряда аниона, а следовательно, и переход его в органическую фазу зависит от
этих параметров. Действительно, если FеСl4- хорошо экстрагируется, то СoСl42- и ZnCl42- совершенно не экстрагируются. Известно, что экстракция галогенидных комплексов металлов возрастает в порядке F- << Cl- < Br- < I-, что отражает увеличение размеров аниона экстрагирующегося соединения.
Как мы уже упоминали, экстракция слабых органических кислот происходит по ассоциативному механизму, и распределяющейся частицей является недиссоциированная ковалентная молекула кислоты. Если же в водной фазе присутствует катион какого-либо металла, а кислота не растворима в воде, то наблюдается экстракция металла в органическую фазу. Уравнение экстракционного процесса можно записать в следующем виде:
![]() ;
;
![]() .
.
Формально эти уравнения полностью совпадают с уравнениями ионообменного процесса на твердой смоле, поэтому такой вид экстракции довольно часто называют ионообменным. В качестве экстрагентов в этом случае используются высшие алифатические карбоновые кислоты с общей формулой RСООН, ароматические сульфо-кислоты R-SO3Н, моно- и диалкилфосфорные кислоты H2RPO4 и HR2PO4. Отличительной особенностью этих систем является полимеризация как самих кислот, так и извлекающихся соединений в органической фазе.